anvio
read recruitment glossary
read here means short pieces of dna, and recruitment here means matching similar short pieces together. it’s like you have a job (a genome), and people (reads) get recruited for it. so, if we have a genome and want to see if it, as a whole or in parts, exists in our sample, we use this technique.
please visit this video to fully grasp the idea.
before doing the read recruitment exercise, here is a glossary that you might find useful:
sequencing
Determining the precise order of nucleotides in a DNA or RNA.
- Sanger sequencing (first generation)
- Next generation sequencing (NGS)
- Third gen (Oxford Nanopore, PacBio SMRT)
reference sequence
- A sequence that serves as the base for comparing and aligning other sequences.
- Anything that is longer than your shorts may serve as a reference.
metagenomes
- Metagenomes are complete or partial genomes that have been gathered from an environment (for example, soil or water).
- They are then sequenced by a sequencing machine to be transferred into a computer.
- The so-called shift from in vitro to in silico.
short reads
- DNA sequences generated during sequencing, ranging from 50 to 300 base pairs.
- They are produced by technologies like Illumina.
- Shorts can be long: you can recruit long reads with your reference sequence, too.
short reads generation
- Fragmentation: Using physical, enzymatic, or chemical methods. These produce fragments of varying size but within a specific range (200–500 base pairs).
- Size selection: Using electrophoresis and purification. Fragments within the desired size range are enriched, while others are discarded.
- Adapter ligation: Short DNA sequences (adapters) are added to the ends of the fragments to attach them to the sequencing platform.
- Sequencing by synthesis: Illumina sequences the fragments. The length is defined by the number of cycles during sequencing.
contig
-
When sequencing generates many reads that overlap, it means that the sequencing machine produces multiple short fragments of DNA that represent the same regions of the original DNA sequence, often with overlapping sections.
What does this mean?- It means that one part of the DNA is being read several times by the machine.
- The parts that are repeated with high likelihood are accepted as correct bases (shorts).
- These shorts are then put together to form a contig, which is longer. You can build a contig with or without a reference sequence.
- Software analyzes the reads to determine where the end of one read matches the start of another. This is how the shorts are assembled in order.
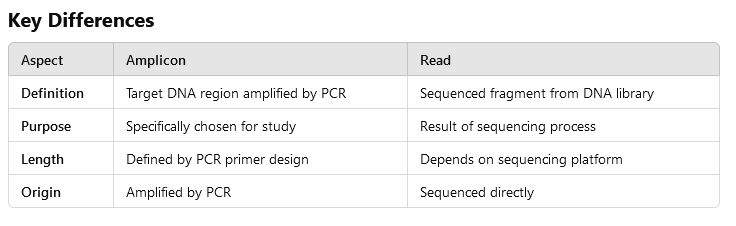
amplicon
- Amplify: To make large; -on: A suffix used in genetics to denote a unit or region.
- A piece of DNA that has been amplified (copied).
- Amplification is often done using PCR to copy those pieces.
- Amplicons are usually small, ranging from 100 to 500 base pairs.
metagenomic
Metagenomics is the study of genetic material recovered directly from environmental samples, bypassing the need to isolate and culture individual organisms.
single copy core genes
- Genes that are typically found as a single copy in the genomes of organisms (bacteria, archaea, or eukaryotes).
MAG
-
Remember how we sliced the DNA into short reads to get it out of the cell? After extracting it, we try to rebuild it. A MAG is what we have rebuilt.
(No, we are not crazy—the technological restrictions dictate this process for now.) - Sometimes we can build the entire genome from the pieces, and other times it is incomplete. Both are called MAGs.
- MAG stands for Metagenome-Assembled Genome.
What is the purpose of read recruitment?
- To investigate one or more reference sequences in the context of one or more samples, which we access through short reads.
- Your reference and short reads can represent any DNA or RNA sequences.
What can serve as a reference?
- Complete genomes (e.g., bacterial, viral, eukaryotic).
- Draft genomes or contigs (incomplete assemblies from sequencing projects).
- Individual genes or regions of interest (e.g., marker genes like 16S rRNA, functional genes).
- Metagenome-assembled genomes (MAGs) from previous analyses.
- Even artificially constructed sequences or hypothetical references.
What can serve as short reads?
Short reads are raw sequencing reads from your dataset. These are obtained by extracting and fragmenting DNA in vitro, sequencing them with a machine, and then analyzing the sequences in silico.
- Metagenomic datasets: Sequences come from a mixed microbial community.
- Transcriptomic datasets: Focus on RNA sequences (e.g., for gene expression studies).
- Amplicon sequencing: Includes sequences like 16S or ITS reads for community profiling.
- Whole-genome sequencing (WGS): Data for a specific organism.
- Even synthetic or simulated reads, depending on the purpose of the study.
Are amplicons connected or separated?
Amplicons can be connected or separated, depending on the context of the analysis:
- Connected: In metagenomics, amplicons can overlap if they are multiple fragments.
- Separated: In PCR or during sequencing and analysis. In Anvio, they are typically treated as separated unless you assemble them into contigs.
What is the difference: amplicons - reads?
The difference between amplicons and reads can be visualized in the following image: