anvio
what is bash and script?
Bash is the language we use to communicate with the terminal (the black tab where we type commands) and script is just a fancy way of saying a file with some lines of commands written in it.
how to save and run a script in bash?
or take a step back: why would we even want to do that?
sometimes we need to run the same commands regularly. instead of retyping or copy-pasting them every time, we can save those commands in a file and simply call that file whenever we need it.
for example in the read recruitment exercise we have a script for looping through the metagenomes for generating a profile.db file for each and we can save that in a file for later use.
so let’s use that example and learn how to save and run it.
first, move to your desired location, like your home directory.
to do that, type cd, and you’ll be out of the current directory. when we don’t specify a directory, it defaults to home.
cd

now we use a tool named nano. we call its name, and it appears. (just type nano and press enter):
nano
nano is like notepad or note app in our phone. it can save some text in it.
then i copy paste this:
names=("batuhan" "alejandra" "jonas" "jessika" "magdalena")
for person in "${names[@]}"; do
echo "Working on ${person} ..."
bowtie2 -x genome -1 metagenomes/${person}-R1.fastq -2 metagenomes/${person}-R2.fastq -S ${person}.sam
samtools view -F 4 -bS ${person}.sam -o ${person}-RAW.bam
samtools sort ${person}-RAW.bam -o ${person}.bam
samtools index ${person}.bam
anvi-profile -i ${person}.bam -c genome.db -o ${person}-profile
rm -rf ${person}.sam ${person}-RAW.bam
done
here we have added a list called “names” and we can change it whenever we want and it makes the script a little bit more general to use.
after pasting it to the nano, we press ctrl O and it asks us to choose a name for our file:
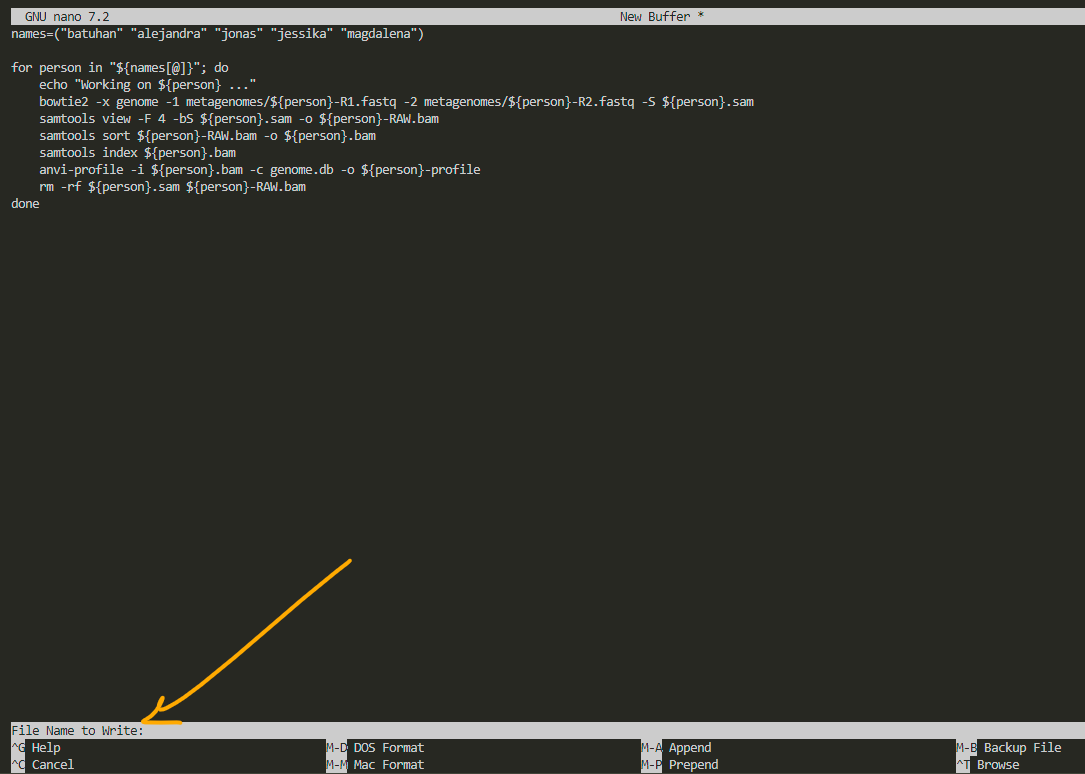
i name it profile.sh to remember that this script creates profiles for visualization. always remember to add .sh as the extension to whatever name you choose. after that, press enter.
for exit, press ctrl X:
now we have the script and we can use it for any other metagenomic data profiling:
for running a .sh file we need to ask bash to run it for us:
bash profile.sh
if we need to run it for other data than these metagenomes, we simply open the script by typing nano profile.sh, edit the list (names), and then run it.
also you’ll see that nano doesn’t like mouse. you need to move around by arrows.